Quick Start Guide (Deutsch)
1. Öffnen des Programms#
Öffnen Sie das Programm in Ihrem Browser, indem Sie auf folgenden Link klicken:
https://www.nmrium.org/nmrium/
2. Öffnen von Spektren#
Um Spektren öffnen zu können, müssen sie in einem der folgenden Dateiformate vorliegen:
- JCAMP-DX (.dx, .jdx, .jcamp)
- Gezippter Ordner im Bruker-Format (Rohdaten oder prozessierte Daten)
- Jeol (.jdf)
- NMRium-Datei (.nmrium)
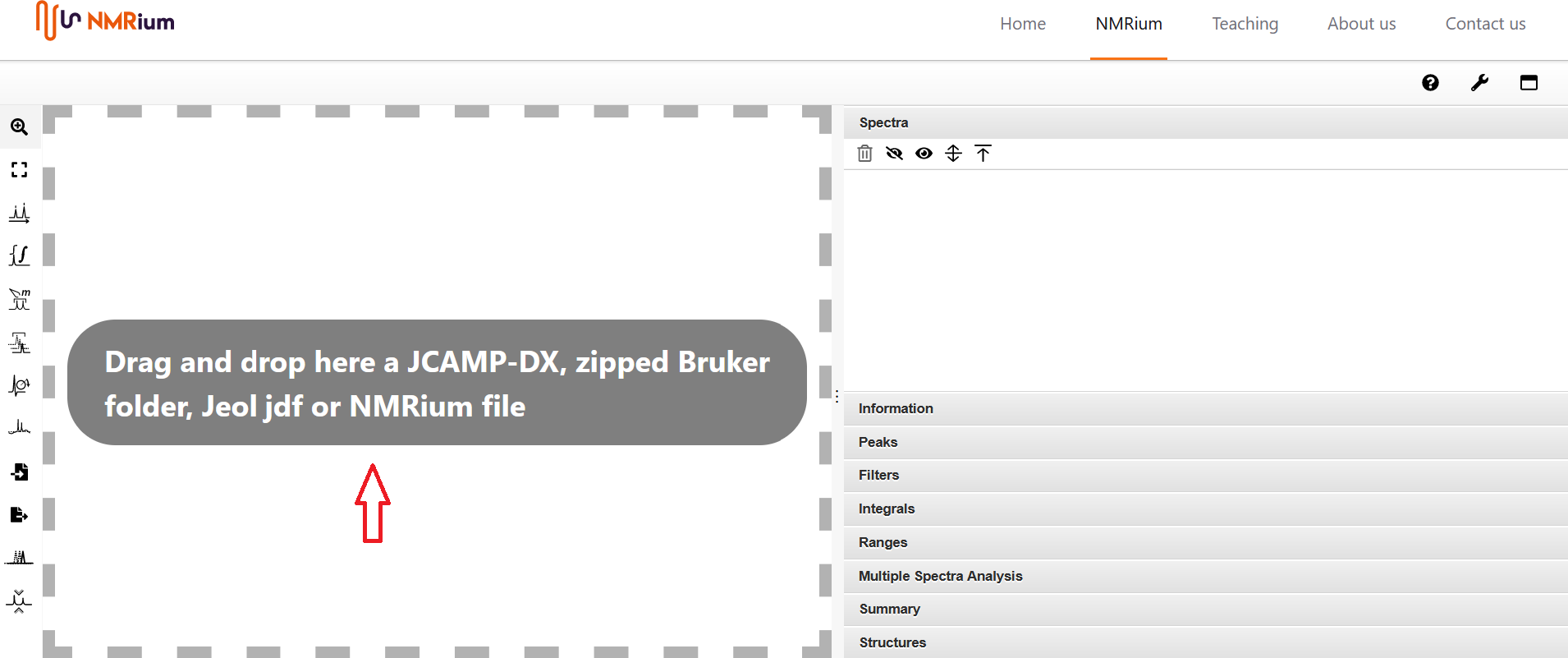
Ziehen Sie die Datei, die Sie öffnen wollen, mit der Maus in die graue Box Drag and drop here in der Mitte des Bildschirms. Sie können ein Spektrenset öffnen, indem Sie entweder die Spektren einzeln in die Arbeitsfläche ziehen oder indem Sie einen gezippten Ordner, der alle Spektren enthält, in die graue Box ziehen. Außerdem können gezippte Bruker-Ordner, die mehrere Spektren enthalten, öffnen.
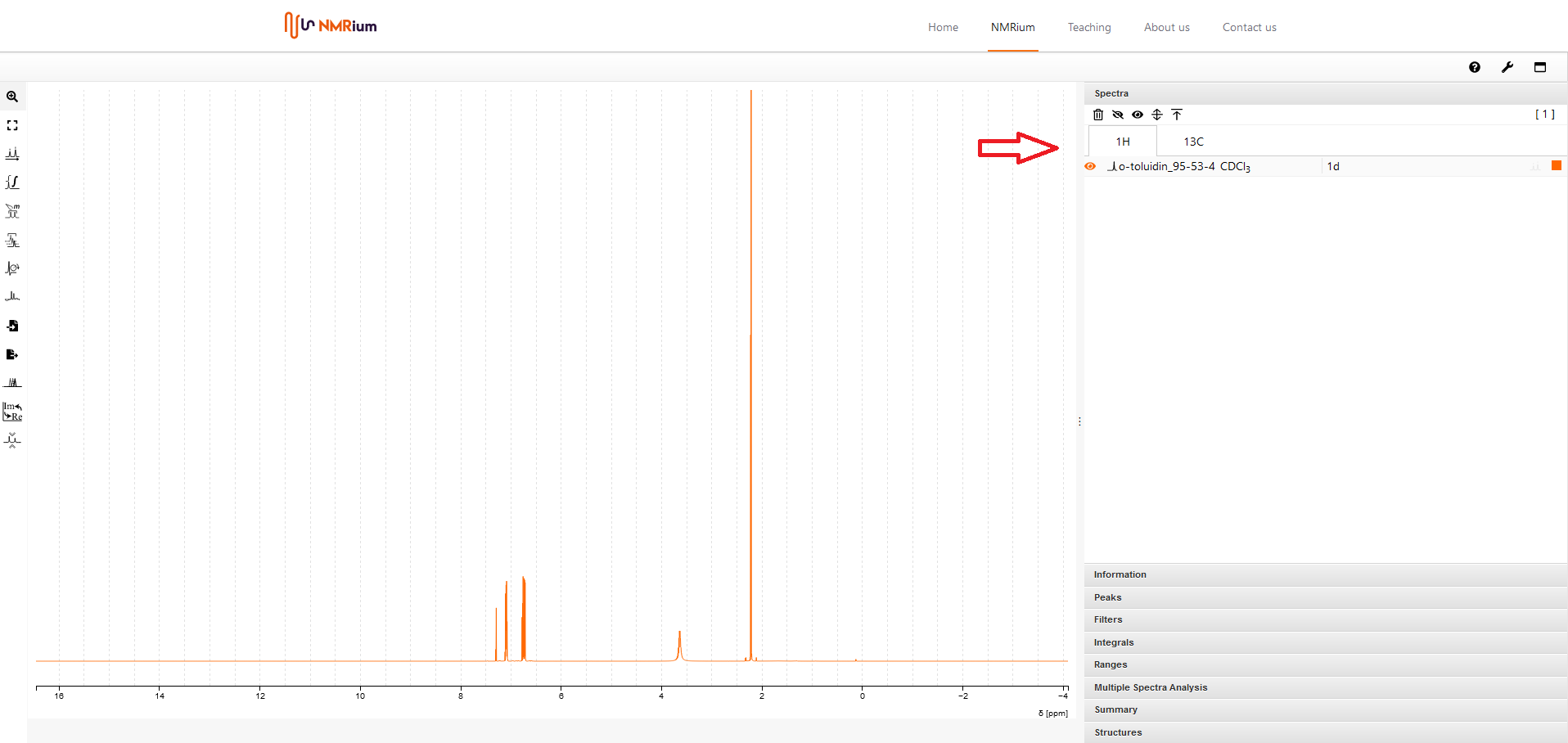
3. Spektren auswählen#
Auf der rechten Seite des Spektrums befindet sich ein aufklappbares Menu mit verschiedenen Panels. Klicken Sie das Panel Spectra an. Die Spektren werden den gemessenen Kernen zugeordnet, zum Beipiel 1H oder 13C etc sowie die 2D-Sprektren den enthaltenen Kernen, zum Beispiel 1H,1H. Wenn Sie auf den entsprechenden Kern klicken, dann finden Sie die dazugehörigen Spektren. Wählen Sie ein Experiment aus, indem Sie auf die entsprechende Zeile klicken. Das Spektrum wird in der Arbeitsfläche geöffnet.
Wobble Kurven
Bruker-Ordner können Dateien mit sogenannten Wobble-Kurven enthalten. Es ist sinnvoll, diese vor der Bearbeitung der restlichen Spektren zu löschen. Sie erkennen Sie an dem Spektrennamen wobble. Klicken Sie das entsprechende Spektrum mit der rechten Maustaste an. Klicken Sie delete in der sich öffnenden Box an, um die Wobble-Kurve zu löschen.
Änderung der Spektrenfarbe
Sie können die Farbe Ihres Spektrums ändern, indem Sie auf den farbigen kleinen Kasten rechts in der Zeile Ihres Spektrums klicken. In dem sich öffnenden Fenster können Sie eine Farbe auswählen. Bei 2D-Experimenten sind die Farben für die positive und die negativen Peaks frei wählbar.
4. Löschen eines Spektrums#
Um ein einzelnes Spektrum zu löschen, klicken Sie mit der rechten Maustaste auf die entsprechende Zeile im Panel Spectra. Klicken Sie anschließend delete, um das Spektrum zu löschen. Wenn Sie alle Spektren eines Kerns löschen wollen, klicken Sie auf das Papierkorbsymbol im Panel Spectra. Klicken Sie anschließend yes im sich öffnenden Fenster.
5. Vergrößern eines Spektrenausschnitts#
Um einen Auschnitt des Spektrums zu vergrößern, fahren sie mit der Maus über den zu vergrößernden Ausschnitt des Spektrums und halten Sie dabei die linke Maustaste gedrückt. Beim Loslassen erscheint der ausgewählte Bereich des Spektrums. Sie können diesen Vorgang beliebig oft wiederholen. Mit einem Doppelklick irgendwo im Spektrum können Sie die Vergrößerung rückgängig machen. Um zur ursprünglichen Vergrößerung zurückzugelangen, klicken sie auf den Button Horizontal zoom out in der Menuebar auf der linken Seite des Spektrums.
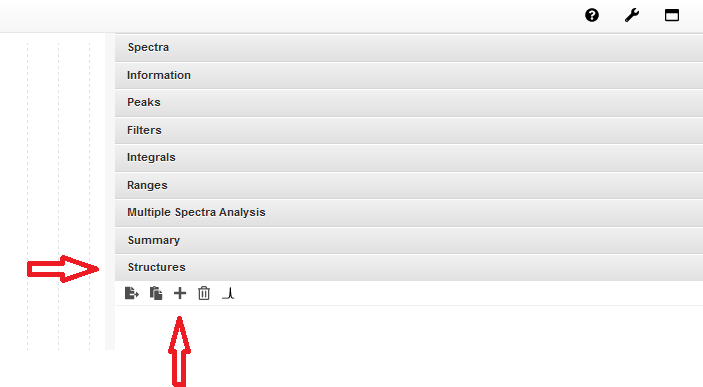
6. Einfügen einer Molekülstruktur#
Es gibt zwei Möglichkeiten, eine Molekülstruktur in das Programm einzufügen:
Molfile
Ziehen Sie eine Molfile-Datei mit der Maus in die Arbeitsfläche. Der Panel Structures wird geöffnet. Dort werden Ihnen sowohl die Molekülstruktur als auch die Summenformel und die molare Massen angezeigt.
Integrierter Struktur-Editor
Klicken Sie auf den Panel Structures auf der rechten Seite des Spektrums und dort anschließend auf den Button +. Es öffnet sich ein Fenster. Dort können Sie Ihre Molekülstruktur selbst zeichnen. Wenn Sie fertig sind, klicken Sie auf Save. Die Struktur wird im Panel Structures gespeichert, Sie finden Sie dort, zusammen mit der Summenformel und der molaren Masse.
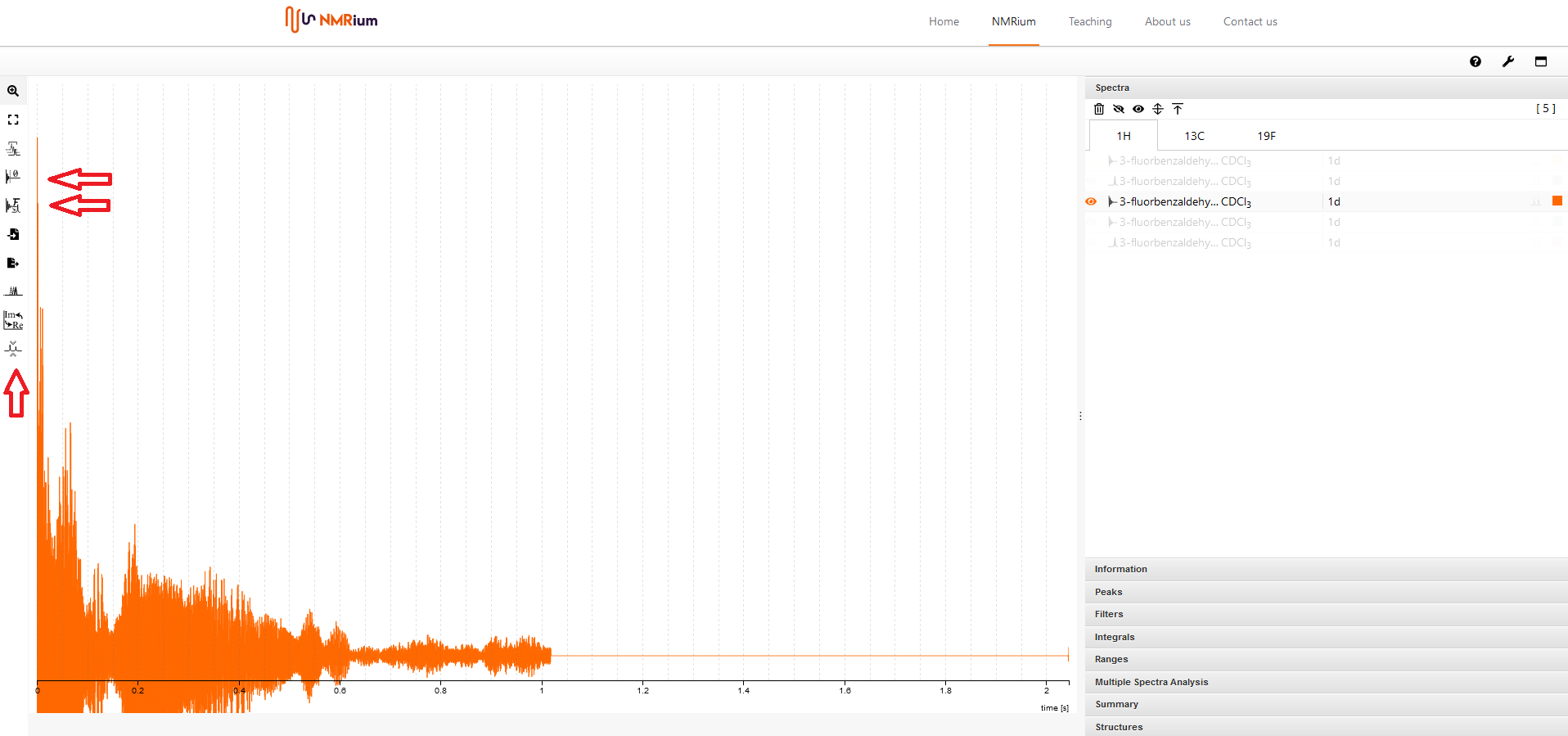
7. Prozessierung der Rohdaten (derzeit ausschließlich 1D)#
NMRium bietet die Möglichkeit, eine Fourier Tranformation (FT) der eindimensionalen Rohdaten durchzuführen. Öffnen Sie den FID, indem Sie auf die entsprechende Datei im Panel Spectra klicjen.
Fourier transformation#
Bevor Sie die Fourier Transformation durchführen, klicken Sie auf den Button Zero Filling in der Menubar auf der linken Seite des Spektrums. Wählen Sie geeignete Werte für die Größen Size (Zero filling) und Line Broading (exponentielle Window Funktion) aus und klicken Sie Apply. Anschließend starten Sie die Fourier Transormation, indem Sie auf den Button FFT Filter in der Menubar auf der linken Seite des Spektrums klicken.
Geeignete Werte für Size und Line Broading
Wenn Sie sich nicht sicher sind, welche Werte Sie verwenden sollen, empfehlen wir Ihnen, folgende Größen anzugeben:
Size: Wählen Sie doppelt so viele Punkte, wie der originale FID hat.
Line Broadening (LB): Geben Sie 0.3 Hz für ¹H-Spektren an und 1-3 Hz für ¹³C-Spektren.
Phasenkorrektur#
Klicken Sie auf den Button Phase correction auf der linken Seite des Spektrums. Sie können dann manuelle Phasenkorrektur und automatische Phasenkorrektur auswählen.
Automatische Phasenkorrektur
Klicken Sie den Button Apply , die Phase wird automatisch korrigiert.
Manuelle Phasenkorrektur
Sie können eine Phasenkorrektur nullter Ordnung und eine Phasenkorrektur erster Ordnung durchführen.
Für die Phasenkorrektur nullter Ordnung klicken Sie den Button PH0 mit der linken Maustaste an und halten Sie ihn gedrückt. Bewegen Sie Maus, bis die Phase korregiert ist, dann lassen Sie die linke Maustaste los. Sie können den Pivot-Punkt (rote Linie) verschieben, indem Sie einen Doppelklick auf dem entsprechenden Signal machen.
Wiederholen Sie den Vorgang mit der Phasenkorrektur erster Ordnung, indem Sie den Button PH1 anklicken und die Maus bewegen. Ihr Fokus sollte hierbei auf den Signalen liegen, die möglichst weit vom Pivot-Punkt entfernt sind. Abschließend klicken Sie den Button Apply, um die Phasenkorrektur abzuschließen.
Basislinienkorrektur#
Klicken Sie auf den Button Baseline correction in der Menubar auf der linken Seite des Spektrums. Sie können zwischen zwei Algorithmen wählen: polynomial und AIR PLS.
- Wenn Sie den Algorithmus polynomial wählen, müssen Sie einen Grad (degree) auswählen und anschließend apply anklicken.
- Wenn Sie den Algorithmus AIR PLS wählen, müssen Sie sowohl ein Maximum als auch einen Toleranzbereich auswählen und anschließend apply anklicken.
8. Festlegung des Referenzwertes#
Klicken Sie den Button Peaks Picking auf der linken Seite des Spektrums an. Anschließend suchen Sie Ihr Lösungsmittelsignal im Spektrum mit der Maus. Wenn Sie das Signal im Fadenkreuz sehen, drücken Sie die Taste shift auf der Tastatur und gleichzeitig die linke Maustaste. Die chemische Verschiebung des Signals wird sowohl im Spektrum über dem Signal als auch im Panel Spectra auf der rechten Seite des Bildschirms angezeigt. Wählen Sie einen der beiden angezeigten Werte aus (im Spektrum klicken Sie mit der linken Maustaste auf den Signalwert, im Panel machen Sie einen Doppelklick) und geben dann den korrekten Referenzwert an.
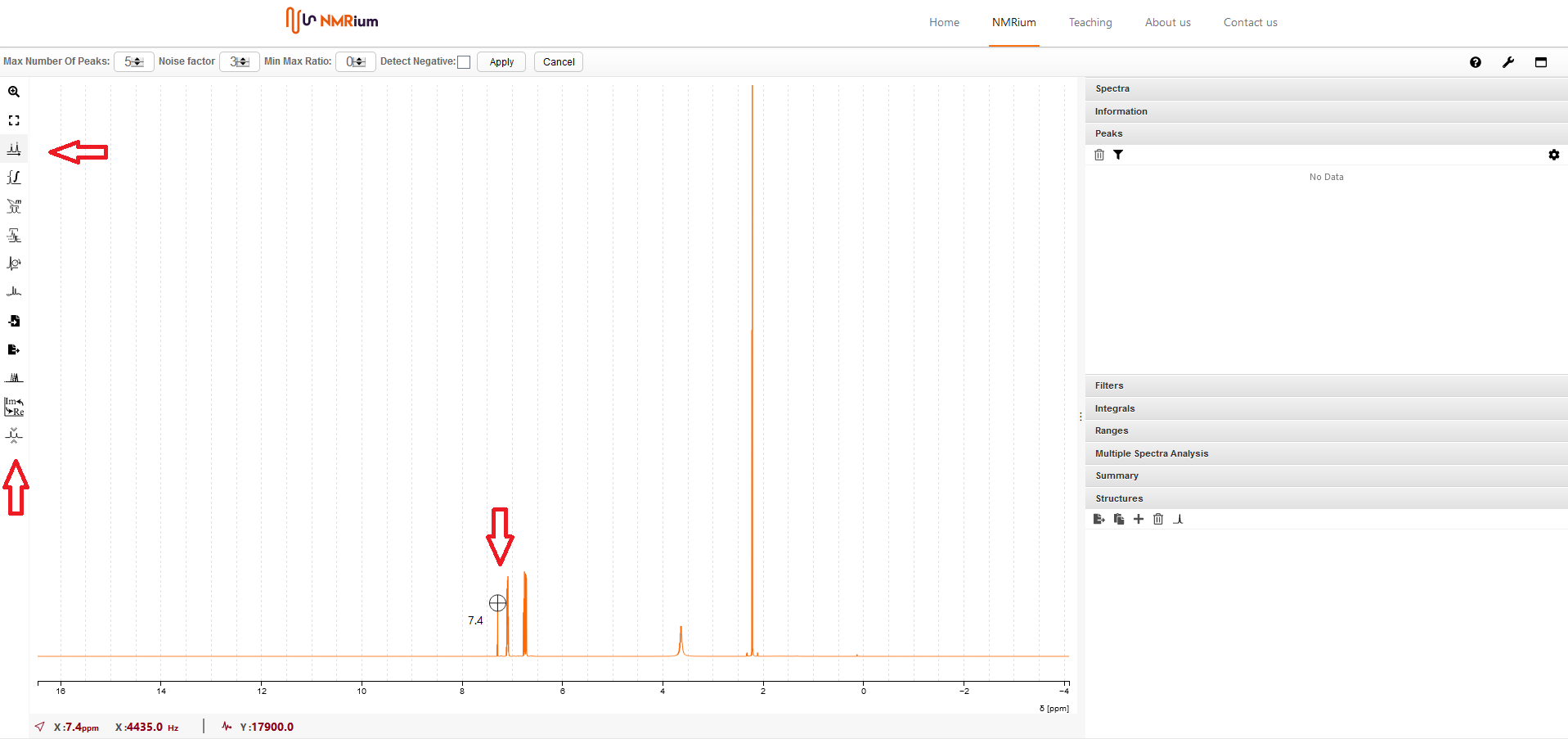
9. Peak Picking#
Klicken Sie den Button PeaksPicking auf der linken Seite in der Menubar an. Sie können ziwschen automatischen Peaks Picking und manuellem Peaks Picking wählen.
Automatisches Peaks picking
In der Zeile oberhalb des Spektrums können Sie die maximale Anzahl an Peaks, den Rausch-Faktor und das Minimum-Maximum-Verhältnis der Peaks (Min Max Ratio) angeben Anschließend klicken Sie auf Apply Die chemischen Verschiebungen werden sowohl im Spektrum über den jeweiligen Signalen als auch im Panel Peaks auf der rechten Seite des Bildschirms in einer Liste angezeigt.
Manuelles Peaks picking
Klicken Sie den Button Peaks Picking auf der linken Seite des Spektrums an. Anschließend suchen Sie Ihr Lösungsmittelsignal im Spektrum mit der Maus. Wenn Sie das Signal im Fadenkreuz sehen, drücken Sie die Taste shift auf der Tastatur und gleichzeitig die linke Maustaste. Die chemische Verschiebung des Signals wird sowohl im Spektrum über dem Signal als auch im Panel Spectra auf der rechten Seite des Bildschirms angezeigt.
10. Löschen von Chemischen Verschiebungen#
Wenn Sie die chemische Verschiebung eines Signals löschen woollen, wählen Sie den entsprechenden Wert im Panel Peaks auf der rechten Seite des Bildschirms. Klicken sie das Papierkorbsymbol auf der rechten Seite der entsprechenden Zeile an, der Wert wird gelöscht..
Wenn Sie alle Chemischen Verschiebungen löschen wollen, klicken Sie das Papierkorbsymbol oberhalb der Liste mit den Chemischen Verschiebungen im Panel Peaks auf der rechten Seite des Bildschirms. Bestätigen Sie, dass Sie alle chemischen Verschiebungen löschen wollen, indem Sie Yes im sich öffnenden Fenster anklicken.
11. Auswahl der Werte, die im Panel Peaks angezeigt werden sollen#
Sie können entscheiden, welche Werte Ihnen im Panel Peaks angezeigt werden. Öffnen Sie hierfür den Panel Peaks auf der rechten Seite des Bildschirms. Klicken Sie auf den Zahnrad-Button. Es werden Ihnen Auswahlmöglichkeiten für alle gemessenen Kerne angezeigt. Sie können zwischen folgenden Werten für jeden Kern auswählen:
- Peak Number (Nummer des Peaks)
- Peak Index (Index des Peaks)
- Chemical shift (Chemische Verschiebung in ppm)
- Chemical shift (Chemische Verschiebung in Hz)
- Width (Breite)
- Intensity (Intensität)
Setzen Sie bei jedem Wert, den Sie angezeigt bekommen wollen, einen Haken. Machen Sie das bei allen angezeigten Kernen. Klicken Sie anschließend auf den grünen Haken oben rechts, um Ihre Einstellungen zu speichern.
12. Integration#
Klicken Sie den Button Integral Tool auf der linken Seite des Spektrums in der Menubar an. Um ein einzelnes Signal zu integrieren, markieren Sie das Signal indem Sie mit der Maus über das Signal fahren, während Sie die Taste Shift auf Ihrer Tastatur drücken und gleichzeitig die linke Maustaste drücken. Machen Sie das mit jedem Signal, das Sie integrieren wollen. Eine Lsite der integrierten Signale finden Sie im Panel Integrals auf der rechten Seite des Bildschirms.
Es ist vorgegeben, dass die relative Anzahl an integrierten Protonen 100 beträgt. Sie können diesen Wert ändern, indem Sie auf das Summensymbol über der Liste im Panel Integrals klicken. Es öffnet sich ein neues Fenster. Geben Sie entweder die korrekte Anzahl an Protonen manuell ein und klicken Sie auf Set oder verwenden Sie die durch die Strukturformel Ihres Moleküls vorgegebene Anzahl durch Anklicken von Set im unteren Bereich des Fensters.
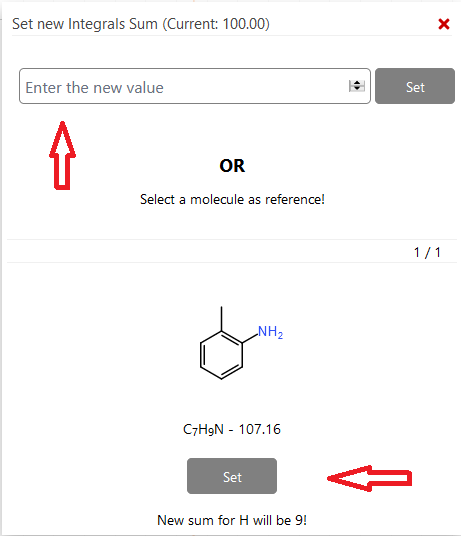
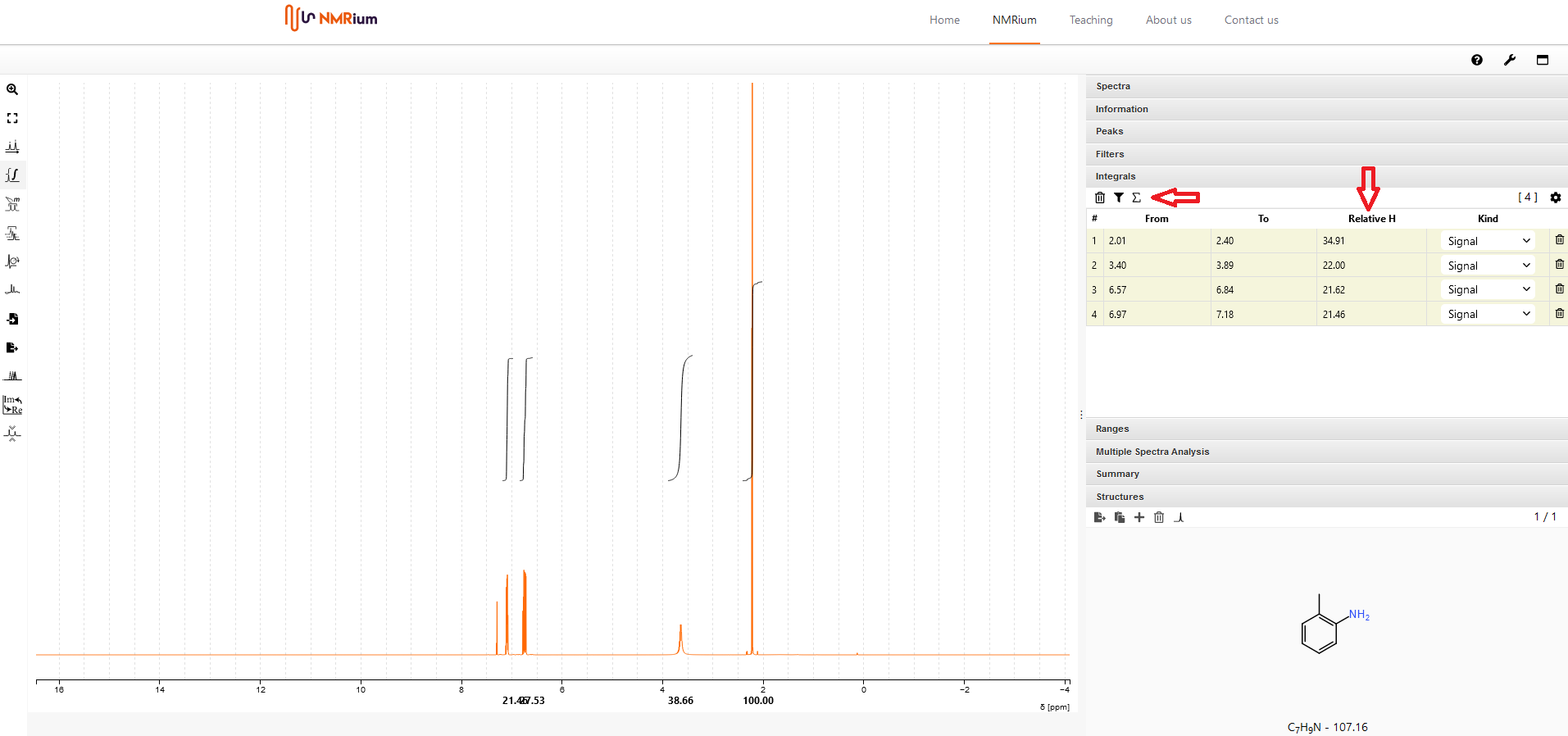
13. Integrale löschen#
Wenn Sie ein einzelnes Integral löschen wollen, wählen Sie dieses im Panel Integrals auf der rechten Seite des Bildschirms aus. Klicken Sie das entsprechende Papierkorbsymbol an, das Integral wird gelöscht. Wenn Sie alle Integrale löschen wollen, klicken Sie das Papierkorbsymbol oberhal der Lsite im Panel Integrals an. Bestätigen Sie, dass Sie alle Integrale löschen wollen, indem Sie Yes im sich öffnenden Fenster anklicken.
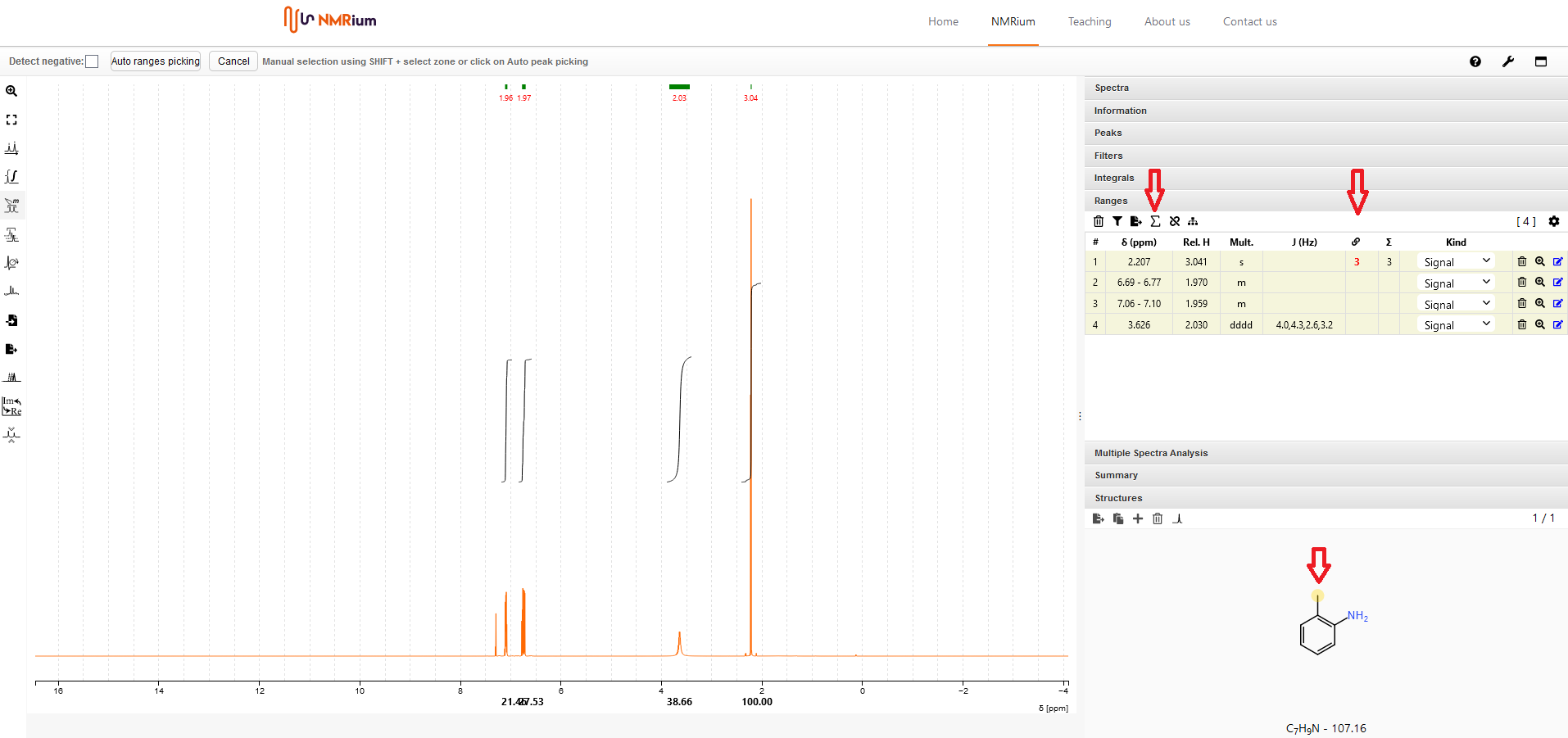
14. Bestimmung von Zuordnungsbereichen und Bestimmung von Kopplungskonstanten#
Klicken Sie auf den Button Ranges Picking auf der linken Seite des Bildschirms in der Menubar. Sie können die Zuordnungsbereiche entweder manuell oder automatisch bestimmen:
Automatische Bestimmung von Zuordnungsbereichen
Klicken Sie auf den Button Auto ranges picking. Die ausgewählten Bereiche werden automatisch bestimmt und sowohl im Spektrum als auch im Panel Ranges auf der rechten Seite des Bildschirms abgebildet. Die dazugehörigen Kopplungskonstanten werden automatisch bestimmt.
Manuelle Bestimmung von Zuordnungsbereichen
Drücken Sie die Shift-Taste und markieren Sie gleichzeitig mit der Maus den Bereich, den Sie als Bereich festlegen möchten. Der markierte Zuordnungsbereich wird sowohl im Spektrum als auch in einer Liste auf der rechten Seite des Spektrums im Panel Ranges angezeigt. Die entsprechenden Kopplungskonstanten werden automatisch ermittelt.
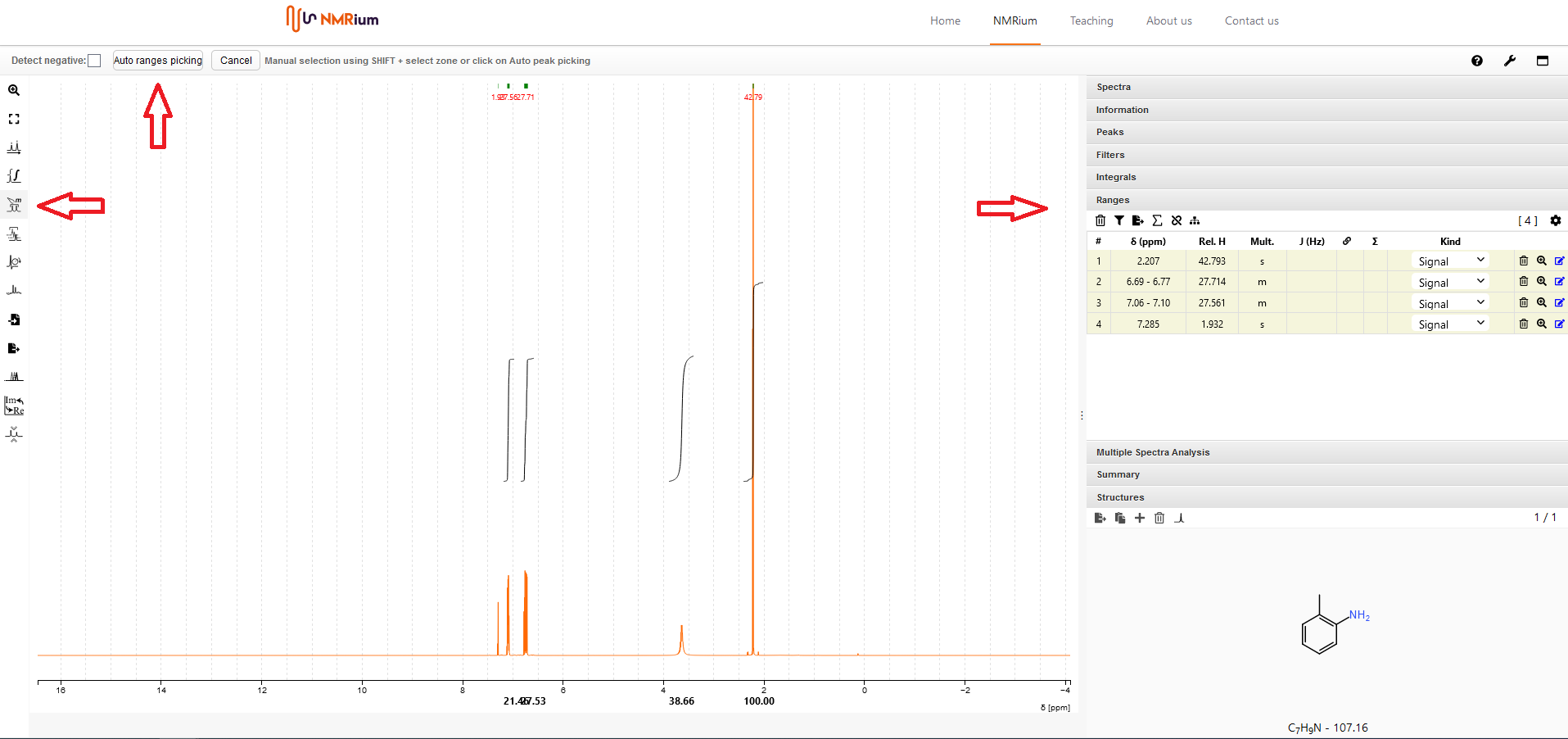
15. Löschen von Zuordnungsbereichen#
Wenn Sie einen einzelnen Zuordnungsbereich löschen möchten, suchen Sie den entsprechenden Bereich auf der rechten Seite des Spektrums in der Liste des Panels Ranges. Klicken Sie auf das Papierkorbsymbol ganz rechts in der Zeile, und der Bereich wird gelöscht. Wenn Sie alle Zuordnungsbereiche löschen möchten, klicken Sie auf das Papierkorbsymbol oberhalb der Werteliste des Panels Ranges. Bestätigen Sie, dass Sie alle Bereiche löschen möchten, indem Sie in dem sich öffnenden Fenster auf Yes klicken.
16. Zuordnung von Signalen#
Um Signale zuzuordnen, müssen Sie Zuordnungsbereiche definieren (Abschnitt 14) und eine Molekularstruktur einfügen (Abschnitt 6). Öffnen Sie die Panels Ranges und Structures auf der rechten Seite des Bildschirms. Geben Sie die korrekte Anzahl der relativen H-Atome an, indem Sie auf das Summensymbol oberhalb der Liste Ranges klicken. In dem sich öffnenden Fenster können Sie entweder die Anzahl der H-Atome manuell eingeben und dann auf Set klicken oder die durch die Summenformel vorgegebene Anzahl der H-Atome übernehmen, indem Sie im unteren Bereich auf Set klicken.
Klicken Sie in der Liste in der Zeile des Signals, das Sie zuordnen möchten, auf das Kästchen unter dem Verknüpfungssymbol. Dort erscheint eine rote Null. Ordnen Sie dann das Signal zu, indem Sie die entsprechenden Atome in der Strukturformel anklicken. Verfahren Sie auf die gleiche Weise mit allen anderen Bereichen. Sie können einem Bereich auch mehrere, unterschiedliche H-Atome zuordnen, indem Sie nacheinander alle H-Atome in der Strukturformel anklicken, die zu dem jeweiligen Signal gehören. Wenn Sie diastereotopische H-Atome zuordnen wollen, halten Sie die Shift-Taste gedrückt, während Sie das entsprechende H-Atom in der Struktur anklicken. Dadurch werden beide diastereotopischen H-Atome sichtbar.
17. Export von zugeordneten Spektren#
Um die zugeordneten Spektren zu exportieren, klicken Sie auf die Buton Export in der Menubar auf der linken Seite des Spektrums. Es öffnet sich ein Feld, in dem Sie auswählen können, in welchem Format Sie Ihr Spektrum exportieren möchten. Sie haben die folgenden Optionen:
- Export as SVG
- Export as PNG
- Save data
- Copy image to Clipboard
Klicken Sie auf Save data, um das zugeordnete Spektrum mit allen ausgewerteten Informationen zu speichern und es später bearbeiten zu können. Es wird eine NMRium-Datei erstellt und zum Download angeboten.
info
Eine NMRium-Datei ist eine komprimierte Textdatei, die alle Informationen der (teilweise) zugeordneten gespeicherten Spektren enthält. Je nach Anzahl der enthaltenen Spektren kann das Speichern länger dauern, ebenso das erneute Öffnen einer NMRium-Datei.